hemofilia
Sinônimos
Hemofilia, distúrbio de sangramento hereditário, deficiência de fator de coagulação do sangue,
Deficiência de fator VIII, deficiência de fator IX, hemofilia
definição
A hemofilia é uma doença hereditária do sistema de coagulação do sangue:
Os pacientes afetados têm coagulação sanguínea prejudicada, que se manifesta no fato de que o sangramento ocorre com o menor trauma e é difícil de estancar.
Os fatores de coagulação não podem ser ativados, de modo que o curso regular da cascata de coagulação não é possível.
A hemofilia é uma doença congênita que se apresenta em duas formas,
Hemofilia A e B, podem estar presentes.
A hemofilia A ocorre com mais frequência (em 85% dos casos) do que a forma B (aproximadamente 15%) e é a mais grave das duas formas.
A atividade do complexo de fator VIII é limitada na hemofilia A porque a formação de fator VIII-C nas células do fígado é reduzida.
O fator VIII-C é uma das duas subunidades do complexo do fator VIII: O von-Willebrand-Fator e fator VIII-C formam este complexo promotor de coagulação na cascata de coagulação. O fator de von Willebrand protege o fator VIII-C de ser decomposto muito rapidamente.
A quantidade de complexo de fator VIII funcional é consequentemente reduzida na forma A.
No hemofilia B. a síntese do fator IX de coagulação ocorre em menor grau, de modo que seu efeito no processo de coagulação é ausente e a hemostasia é prejudicada.
Ocorrência / epidemiologia
A ocorrência de hemofilia na população é para hemofilia A. 1 em 5.000 e para a forma B. 1:15.000.
O fato de os homens, em particular, ficarem doentes se deve a questões relacionadas ao gênero
Forma de herança ligada ao X do defeito de coagulação (ver causa / desenvolvimento).
Fundamentos
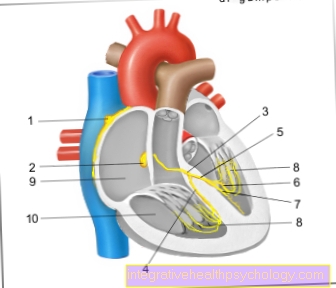
A coagulação do sangue é um sistema bem ajustado: vários componentes do sangue, os chamados fatores de coagulação, são ativados um após o outro em forma de cascata para permitir a hemostasia em caso de lesão.
O sistema de coagulação consiste na hemostasia primária e secundária, também chamada de coagulação plasmática.
Se a parede de um vaso é lesada, o vaso afetado se estreita em uma primeira etapa (= coagulação primária) Em seguida, um suporte (=Trombo de plaquetas) na área lesada, que é coberta por plaquetas (= Plaquetas) é formado a fim de obter uma vedação inicial do vaso.
A formação do trombo ativa a coagulação plasmática:
A cascata de coagulação começa e a interação dos vários fatores de coagulação cria uma ferida estável ou fechamento vascular.
Causa e origem
o hemofilia Ambas as formas de hemofilia são herdadas como traço recessivo ligado ao X.
No chamado modo de herança ligado ao X da hemofilia, a informação genética para um produto gênico está no cromossomo sexual X, que é encontrado em números duplos nas mulheres e em números únicos nos homens.
Existem dois genes para cada informação genética, os chamados alelos. Se houver herança recessiva, ambos os alelos devem ser afetados por uma mutação para causar a doença: Recessivo significa que a informação genética alterada patologicamente é derivada de um saudável, ou seja, não muda, a informação é suprimida e não leva a doenças. Uma doença hereditária recessiva só se desenvolve se ambos os alelos carregam informações genéticas incorretas.
É apenas uma A expressão do gene mudou, vamos Não para hemofilia.
Informação: herança
Se apenas uma expressão de gene for alterada, não haverá doença.
Uma vez que os homens têm apenas um cromossomo X no par de cromossomos sexuais e, portanto, as informações para produtos genéticos em seu cromossomo X estão disponíveis apenas de forma simples, eles ficarão doentes se apenas um alelo for defeituoso.
As mulheres, por outro lado, que têm dois cromossomos X como cromossomos sexuais, só ficam doentes quando ambos os cromossomos têm genes defeituosos para o produto do gene.
Em homens que sofrem de hemofilia, o alelo que carrega a informação hereditária para o fator de coagulação VIII-C (hemofilia A) ou fator de coagulação IX (hemofilia B) está disponível apenas em uma forma ligeiramente modificada;
As mulheres precisam carregar essa mudança genética em ambos os alelos para ficarem doentes.
Se as mulheres têm um alelo defeituoso e um não alterado, são chamadas de portadoras de uma doença hereditária ligada ao X, como a hemofilia.
Na hemofilia, a informação genética defeituosa tem o efeito de reduzir a atividade do fator de coagulação afetado ou de reduzir a produção do fator de coagulação.
O cromossomo X é o portador do material genético dos fatores de coagulação para hemostasia secundária.
Aproximadamente. 70% das mutações no material genético que causam a hemofilia são transmitidas nas famílias, 30% das doenças surgem de alterações espontâneas (= mutações espontâneas) na informação genética.
diagnóstico
Depois de perguntar sobre o histórico médico do paciente e fazer um exame físico completo, existem etapas adicionais no diagnóstico de hemofilia:
Em 2/3 dos casos, há casos de hemofilia na família, razão pela qual um agrupamento familiar da doença deve ser questionado se o paciente se apresenta ao médico com sintomas suspeitos de hemofilia.
Os pacientes relatam hematomas como resultado dos menores ferimentos.
Um diferencia aqueles Gravidade da hemofilia em uma forma leve, moderada ou grave da doença.
O exame de uma amostra de sangue fornece os seguintes resultados típicos para hemofilia:
O tempo de sangramento é normal (= a coagulação primária está intacta), mas a função da coagulação plasmática é reduzida, por isso o chamado tempo de PTT é maior.
PTT significa o tempo parcial de tromboplastina. É medido em testes de laboratório para verificar a função dos fatores I, II, V, VIII a XII e também XIV e XV.
Como um fator na cascata de coagulação está faltando na hemofilia, ela não pode funcionar de maneira ideal, o que resulta em um tempo de coagulação prolongado.
Para ser capaz de diferenciar entre hemofilia A e B, o sangue do paciente deve ser examinado para os fatores VIII e IX; a ausência dos dois fatores determina o tipo de hemofilia.
Diagnóstico diferencial
A hemofilia deve ser diferenciada de outras doenças do sistema de coagulação, sendo mencionada em particular a síndrome de von Willebrand-Jürgens.
O fator de von Willebrand (vWF) trabalha em conjunto com o fator VIII-C im
O fator VIII complexo e provoca a inibição da degradação prematura do fator VIII, além disso, o fator promove a coagulação por mediar a adesão das plaquetas sanguíneas ao sítio vascular lesado. Portanto, o FvW é uma parte importante da coagulação primária e secundária.
Se a síndrome de von Willebrand-Jürgens estiver presente, então a formação do
Os vWFs nas células da parede do vaso ocorrem apenas em pequena extensão ou de maneira incorreta. A redução da taxa de educação ou o funcionamento inadequado do
O fator de von Willebrand é causado por mutações no gene para a formação do fator. A herança da síndrome ou da mutação do gene causador é herdada de maneira autossômica dominante: a informação genética para o FvW está no cromossomo número 12, que não é um cromossomo sexual. No modo de herança dominante, um alelo doente suprime o efeito do segundo alelo saudável, de modo que a doença é causada pela mutação de um gene e causa sintomas clínicos.
Os pacientes geralmente sofrem de sangramento da pele e da membrana mucosa, menos de sangramento espontâneo como os pacientes com hemofilia.
A doença geralmente só é percebida quando há sangramento prolongado após uma operação.
O tempo de sangramento do paciente após uma lesão é prolongado e os valores da coagulação plasmática são alterados patologicamente (PTT prolongado).
A síndrome de von Willebrand-Jürgens é causada pela administração de desmopressina ou pela substituição do fator VIII e do FvW para estabilizar a coagulação (para uma explicação mais detalhada, consulte "Terapia da hemofilia").
Quais são os sintomas da hemofilia?
Os sintomas nas formas de hemofilia não diferem:
- A hemofilia geralmente leva apenas a sintomas clínicos em homens.
- O sangramento excessivo ocorre com a hemofilia, o que não tem relação com o acidente anterior, principalmente banal (trauma), o tempo de sangramento é normal, mas o sangramento normalmente ocorre, o que não ocorre em pessoas saudáveis.
- Uma distinção pode ser feita entre três formas de hemofilia, que são definidas pela atividade residual dos fatores de coagulação afetados:
1hemofilia grave (hemofilia) com menos de 1% de atividade residual ou uma proporção do fator funcional ainda presente em que ocorre sangramento espontâneo e sangramento articular
2. Hemofilia moderada (doença do sangue) com atividade de fator variando entre
1 e 5% da atividade do fator normal e a ocorrência de hematomas (= hematomas) após pequenos traumas
3. hemofilia leve (hemofilia) em que ainda há atividade residual de 5 a 15% e em que os pacientes relatam sintomas como hematomas (hematomas) após trauma significativo e sangramento pós-operatório.
Leia mais sobre o assunto: Quais são as causas de uma hemorragia cerebral
- Mulheres que geralmente têm apenas informações genéticas alteradas para hemofilia geralmente não apresentam sintomas clínicos com mais de 50% de atividade residual.
- Os pacientes apresentam sangramento em grandes articulações como essa joelho-, Quadril- ou Articulação do ombro, onde se fala de hemartrose. O sangramento desencadeia uma reação inflamatória com processos de reparo na articulação, que podem levar ao enrijecimento da articulação.
- Ele também pode sangrar no Musculatura e os tecidos moles vêm. O sangramento leva a um aumento da pressão nos músculos ou no tecido, já que agora há mais volume. Isso pode afetar especialmente seus braços e pernas Síndrome do compartimento liderar:
O aumento da pressão causa a compressão dos vasos sanguíneos e nervos, de modo que as extremidades ficam sem suprimento e grandes áreas de tecido podem morrer. A síndrome compartimental precisa ser tratada pelo cirurgião o mais rápido possível para evitar a perda da extremidade. - Ocorre sangramento no abdome, o que é uma situação de risco de vida para o paciente.
- Após a cirurgia, é possível ocorrer sangramento de longa duração incomum.
Além disso, pode haver longos períodos com sangue na urina. Aqui se ameaça Anemiaporque o paciente está constantemente perdendo sangue, possivelmente despercebido. - São particularmente perigosos Hemorragia cerebral (= sangramento intracraniano), que leva à morte em 10% das pessoas com hemofilia. Você pode descobrir mais sobre este tópico em nosso tópico Hemorragia cerebral.
Como é realizada a terapia para hemofilia?

O sangramento deve ocorrer no doente hemofilia deve ser evitado, razão pela qual o paciente não Medicamento que inibem a coagulação do sangue, como Ácido acetilsalicílico (Aspirin®) e sem injeções intramusculares (= seringas no Musculatura), respectivamente.
Se ocorrer um trauma com sangramento, uma hemostasia local cuidadosa é de grande importância para evitar o sangramento nos tecidos vizinhos.
Terapia medicamentosa do hemofilia consiste na reposição dos fatores de coagulação que o organismo não consegue produzir em quantidade suficiente.
Pacientes com um ligeiro hemofilia receber as preparações de fator de coagulação conforme necessário, ou seja, se houve trauma com sangramento ou se uma grande cirurgia está planejada, que pode resultar em sangramento abundante.
A reposição profilática de fator deve ser usada em pacientes com hemofilia moderada a grave, ou seja, o fator que falta deve ser dado permanentemente, antes sangramento ocorre.
Se houver atividade residual do fator VIII de mais de 15% ou do fator IX de mais de 20-25%, a terapia regular não é necessária; esses pacientes recebem o fator de coagulação ausente em caso de sangramento espontâneo ou antes da cirurgia planejada.
A terapia de longo prazo, bem como a anterior à cirurgia, pode assumir a forma de autotratamento domiciliar, no qual o próprio paciente aplica o fator de coagulação ausente.
Há uma alternativa de terapia para pacientes com hemofilia leve:
A substância ativa Desmopressina (por exemplo, Minirin®) leva a uma distribuição do
Fator VIII, que é armazenado nas paredes dos vasos.
No entanto, o medicamento só pode ser administrado por alguns dias de cada vez, pois, após o estímulo da liberação do fator, a memória nas paredes dos vasos se esgota e precisa ser replicada novamente.
Na terapia aguda, o hemofilia existem três opções de intervenção:
- O paciente recebe protrombina ativada, uma substância que promove a coagulação do sangue para que o sangue seja interrompido o mais rápido possível.
- Outra opção terapêutica é a administração de preparações de fator VII. Este fator está no início da cascata de coagulação e inicia seu curso regular.
- A terceira opção terapêutica é a aplicação de fator animal VIII-C para permitir ao paciente estancar o sangramento.
Complicações
O presente substituto (=substituição) de fatores de coagulação pode levar à formação de anticorpos justamente contra esses fatores, de modo que a substituição em uma dose constante não tem mais efeitos terapêuticos, por isso se fala em formação de inibidores.
Dependendo da determinação da concentração de anticorpos no sangue do paciente, podem ser administradas altas doses de fator VIII, cujo objetivo é recuperar a tolerância a esse fator e interromper a formação de anticorpos. Este procedimento só deve ser realizado em centros especializados.
Existe um baixo risco de contrair hepatite ou contrair o vírus HIV (=HIV), pois os fatores são derivados de produtos do sangue humano.
Outro risco da terapia de substituição para hemofilia é a ocorrência de reações alérgicas.