Doenças genéticas
definição
Uma doença genética ou doença hereditária é uma doença causada por um ou mais genes da pessoa em questão. O DNA atua aqui como um gatilho direto da doença. Para a maioria das doenças genéticas, as localizações dos genes causadores são conhecidas. Em caso de suspeita de doença genética, o respetivo diagnóstico pode ser feito através de exame genético.
Por outro lado, há também um grande número de doenças cuja ocorrência tem influência genética ou é discutida, como diabetes mellitus (“diabetes”), osteoporose ou depressão. Estas são as chamadas disposições, ou seja, uma probabilidade aumentada de certas doenças. As disposições devem ser distinguidas das doenças hereditárias.

Estas são doenças hereditárias comuns
Em termos absolutos, as doenças hereditárias não são comuns, mas as doenças hereditárias listadas aqui ocorrem com frequência em comparação com outras doenças de causa genética.
-
Síndrome de Marfan
-
Anemia falciforme
-
Hemofilia (hemofilia A ou B)
-
Mutação do fator V Leiden e resistência APC resultante
-
Fraqueza Vermelho Verde
-
Deficiência de glicose-6-fosfato desidrogenase (deficiência de G6PD)
-
Polidactilia ("dedos múltiplos", também possível como sintoma em outras doenças)
-
Trissomia 21 (síndrome de Down)
-
Chorea Huntington
causas
As doenças hereditárias são extremamente diversas em sua aparência. Basicamente, eles têm apenas uma coisa em comum: a causa de cada um deles está no DNA, ou seja, no material genético da pessoa em questão. Várias mudanças podem ocorrer aqui, como mutações (troca de informações de DNA) ou deleções (falta de determinado material genético).
Uma grande quantidade de informações é codificada no material genético, como as “plantas” de vários componentes importantes para o funcionamento de uma célula corporal. Podem ser enzimas, canais eletrolíticos ou substâncias mensageiras, por exemplo. Esses menores elementos são então lidos incorretamente ou não são lidos no DNA, que então está ausente no sofisticado sistema do corpo. A informação genética errada ou ausente, portanto, causa certos problemas de funcionamento no corpo. Estes então causam sintomas de acordo com o sistema funcional no qual um elemento está faltando.
Descubra tudo sobre o assunto aqui: O teste genético.
É assim que as doenças hereditárias são herdadas
Todas as doenças hereditárias são herdadas monogeneticamente ou poligeneticamente: isso significa que há uma ou mais localizações genéticas que precisam ser alteradas para levar a uma doença.
Além disso, os traços genéticos sempre podem ser herdados de maneira dominante ou recessiva: Recessivo significa que deve haver uma predisposição para essa doença hereditária específica nos genes paternos e maternos. No caso de herança dominante, uma mudança (ou seja, um dos pais) é suficiente para desencadear a doença. Segue-se que com doenças hereditárias dominantes, as pessoas que são portadoras também ficarão doentes - enquanto com uma herança recessiva geralmente nem se sabe que uma predisposição genética correspondente está presente.
Existem também doenças que são herdadas por meio dos cromossomos sexuais, como hemofilia ou cegueira vermelho-verde. As instalações para isso geralmente estão no cromossomo X, uma vez que o cromossomo Y é muito pequeno e geralmente pode armazenar pouca informação genética. Portanto, fala-se de doenças hereditárias ligadas ao X. Geralmente afetam significativamente mais homens do que mulheres, já que as mulheres podem compensar qualquer informação incorreta no cromossomo X com o segundo.
Em geral, é fácil pesquisar como exatamente uma doença genética é herdada se você estiver interessado.
Testes antes do nascimento
Em princípio, o material genético da criança já pode ser examinado no útero para todas as doenças hereditárias cujas localizações genéticas causais são conhecidas. No entanto, as análises genéticas são demoradas, por isso normalmente apenas se analisa a localização suspeita do gene - para isso, por sua vez, deve haver uma suspeita justificada de uma doença genética.
Para tal exame, o material genético pode ser retirado do líquido amniótico ou da placenta e usado para a análise.
No entanto, deve-se sempre ter em mente que qualquer diagnóstico invasivo também envolve um risco para a vida do feto. Portanto, esses furos devem ser pesados individualmente em cada caso.
Existem também medições que podem indicar uma doença genética, como a medição da transparência nucal como um sinal de trissomia 21. Esses métodos não são perigosos para o feto, mas não podem oferecer certeza absoluta sobre a presença de uma doença genética. Portanto, aqui também, uma operação deve ser cuidadosamente considerada.
Trissomia 21
A causa da trissomia do cromossomo 21 é o cromossomo 21, que não está presente duas, mas três vezes nas pessoas afetadas. Esta variante do DNA é criada quando os cromossomos são distribuídos nas células germinativas parentais, isto é, os espermatozoides ou óvulos. É, portanto, um "erro de distribuição" e não uma mudança no material genético real. Isso explica por que a trissomia 21 pode ocorrer espontaneamente em todas as famílias e por que a probabilidade de ter um filho com síndrome de Down é a mesma em todas as famílias. Estritamente falando, a trissomia 21 - como outras trissomias - não deve ser contada como uma doença hereditária no verdadeiro sentido. No entanto, a trissomia do 21 é a doença relacionada ao DNA mais comum em recém-nascidos.
As características do conjunto alterado de cromossomos na síndrome de Down já podem ser vistas no feto no útero: atrasos e defeitos no crescimento podem levar a, entre outras coisas, um crânio muito pequeno, ossos curtos da coxa e do braço e defeitos cardíacos. Uma grande quantidade de líquido amniótico também pode ser uma indicação de trissomia do cromossomo 21, uma vez que o nascituro afetado bebe ou engole relativamente pouco líquido amniótico. No entanto, nenhuma dessas características é um sinal definitivo da síndrome de Down!
Além dos sinais de retardo de crescimento mencionados, as crianças com síndrome de Down frequentemente também apresentam atraso no desenvolvimento, por exemplo, nas áreas de linguagem e habilidades motoras. Pessoas afetadas pela síndrome de Down costumam mostrar habilidades sociais notáveis, enquanto a inteligência frequentemente permanece abaixo da média. No entanto, as pessoas afetadas diferem muito nessas características, e não é incomum que se formem na escola depois de receber um bom suporte.
Mais tarde na vida, as pessoas com trissomia do cromossomo 21 têm um risco aumentado de serem diagnosticadas com certas doenças. Estes incluem a doença de Alzheimer, epilepsia e câncer, particularmente leucemia. No entanto, a expectativa de vida das pessoas com síndrome de Down continua a aumentar: Nesse ínterim, as pessoas afetadas geralmente chegam aos 60 ou 70 anos.
Você pode encontrar mais informações em nosso site Síndrome de Down
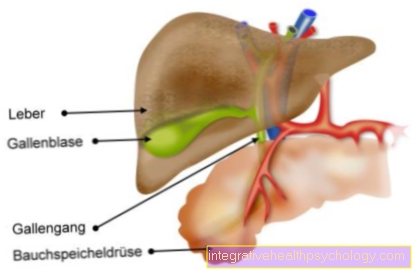
Deficiência de alfa-1 antitripsina
A deficiência de alfa-1 antitripsina pode assumir diferentes formas e formas, dependendo das características genéticas exatas da pessoa afetada. Isso significa que nem toda deficiência de alfa-1 antitripsina leva a sintomas. A seguir, apenas o tipo clinicamente conspícuo (PiZZ) desta doença geneticamente determinada será discutido.
O defeito enzimático presente nesta doença causa a degradação e a remodelação dos blocos de construção no tecido do órgão nas pessoas afetadas. Além disso, as proteínas defeituosas são filtradas do sangue pelo fígado e aí se acumulam. Isso pode causar inflamação do fígado (hepatite), cirrose ou câncer de fígado. As vias aéreas nos pulmões tornam-se instáveis devido à falta de tecido estável e colapsam mais rapidamente: O quadro clínico de DPOC (doença pulmonar obstrutiva crônica) se desenvolve. Este quadro clínico costuma ser o primeiro sintoma de deficiência de alfa-1 antitripsina, portanto, qualquer pessoa com DPOC em uma idade mais jovem deve ser verificada quanto à deficiência de alfa-1 antitripsina.
Se a doença persistir por muito tempo, os pulmões podem inflar excessivamente, pois o ar que você respira não pode ser expirado adequadamente pelas vias respiratórias instáveis e se acumula nos pulmões. Como terapia, além de evitar o tabagismo de forma consistente e vacinações regulares para prevenir doenças respiratórias, medidas medicamentosas também devem ser tomadas: A alfa-1-antitripsina ausente pode ser administrada por via intravenosa para aliviar os sintomas tanto quanto possível e interromper o curso da doença.
Você pode encontrar mais informações em nosso site Deficiência de alfa-1 antitripsina
hemofilia
O grupo da hemofilia também é conhecido coloquialmente como “hemofilia”, pois esse termo descreve com muita precisão o principal sintoma dessa doença hereditária: as pessoas afetadas sangram por mais tempo e, dependendo da gravidade da doença, mais frequentemente do que não afetadas.
O sangramento geralmente é interrompido pelo que é conhecido como cascata de coagulação, uma via de sinalização endógena que evita a perda excessiva de sangue. Neste sistema de coagulação, 13 fatores desempenham um papel, que se ativam um após o outro. Isso pode ser imaginado como uma série de dominós: se você acertar uma pedra (fator de coagulação), ela ativa a próxima, e assim por diante. No final desse caminho de sinal ou do dominó, há coagulação do sangue. Com a hemofilia, um certo fator está faltando - dependendo do subtipo específico da doença: a reação em cadeia é interrompida aqui.
A terapia para a doença pode ser realizada determinando o fator ausente e adicionando-o de fora. As pessoas afetadas devem, portanto, injetar-se regularmente com uma preparação com esse fator de coagulação, para que o resto da reação em cadeia possa ocorrer.
Você pode encontrar mais informações em nosso site Doença de sangue
Fibrose cística
Na doença genética fibrose cística - também conhecida como fibrose cística - há uma produção defeituosa de canais iônicos, mais precisamente de canais de cloreto. Como resultado, a composição das secreções corporais (por exemplo, suor, secreções do trato respiratório e do pâncreas) das pessoas afetadas é alterada: uma vez que a falta de cloreto significa que menos água é puxada para o ducto da respectiva glândula, a secreção é relativamente viscosa.
Como resultado, os sintomas geralmente se desenvolvem no trato digestivo, já que a secreção com as enzimas digestivas não pode fluir bem do pâncreas para o intestino e, portanto, danifica o próprio pâncreas. Além disso, distúrbios digestivos, como fezes gordurosas, diarréia e o baixo peso corporal resultante são comuns.
O segundo grande grupo de sintomas geralmente se desenvolve nos pulmões: como o muco que ocorre naturalmente nos pulmões é mais viscoso do que em pessoas saudáveis, é mais difícil removê-lo dos cílios. Isso pode causar tosse crônica e bloqueio dos brônquios (bronquiectasia). A maior quantidade de secreção pulmonar também proporciona um bom ambiente para o crescimento de bactérias, o que resulta em infecções respiratórias frequentes e pneumonia.
A fibrose cística é tratada sintomaticamente com expectorantes, enzimas digestivas e antibióticos para infecções.
Você pode encontrar mais sobre isso em nosso site Fibrose cística
Fator V Leiden e resistência APC
Uma mutação do fator V Leiden envolve uma alteração na informação genética que pode causar aumento da coagulação do sangue. A razão para isso é o fator V na chamada cascata de coagulação do corpo: esse caminho de sinal garante que, em caso de lesão, a ferida seja fechada pelas próprias "proteínas adesivas" do corpo (fibrina). Existem 13 fatores neste caminho de sinalização, que são nomeados com algarismos romanos (significa “Fator 5 sofrimento”!). O fator V tem um efeito benéfico na formação de um tampão de fibrina, mas também pode ser inibido pela chamada proteína C ativada (APC). Isso desempenha um papel importante na regulação dessa via de sinalização e na prevenção da coagulação sanguínea excessiva.
O fator V mutado está presente nos indivíduos afetados, mas não responde ao APC. O corpo carece de um importante "dispositivo de segurança" neste ponto para evitar a coagulação do sangue sem motivo, o que pode até bloquear os vasos e, assim, causar distúrbios circulatórios.
Estatisticamente falando, as pessoas que são afetadas por uma mutação do fator V Leiden são mais propensas a experimentar um evento trombótico (ou seja, uma trombose ou embolia pulmonar), mesmo sem uma história de fatores de risco típicos. Em termos técnicos, também se fala em “trombofilia”, ou seja, tendência à coagulação.
Você pode encontrar mais sobre isso em nosso site Fator V Leiden
Doença de Gaucher
Na doença de Gaucher, a mudança na informação do DNA causa um defeito em uma enzima envolvida no metabolismo lipídico, mais precisamente a glucocerebrosidase: isso ajuda a quebrar os componentes das células antigas. Em caso de defeito, pode ocorrer uma redução da funcionalidade ou mesmo uma perda de funcionalidade e, consequentemente, os sintomas aparecem na infância ou na idade adulta jovem.
Os sintomas da doença de Gaucher se devem em grande parte ao aumento do fígado e do baço, cujo crescimento o corpo tenta compensar pela falta de enzimas. Isso aumenta a degradação de todos os componentes do sangue, que podem ser reconhecidos no hemograma e usados como um indicador de diagnóstico junto com o aumento do fígado e do baço.
A enzima glicocerebrosidase ausente pode ser usada terapeuticamente como um medicamento. O prognóstico e o curso da doença de Gaucher dependem muito da gravidade da perda de função da enzima.
Para mais informações, leia aqui: Doença de Gaucher.
Doença de Osler
A doença de Osler é uma doença hereditária que se caracteriza por uma forte vasodilatação. Em princípio, essa expansão dos vasos pode ocorrer em qualquer lugar, tanto na pele quanto nos órgãos internos. As paredes dos vasos aumentados são relativamente finas e rasgam facilmente. Como resultado, as áreas afetadas sangram rapidamente.
A vasodilatação ocorre com particular freqüência na face e na membrana mucosa nasal, de modo que as pessoas afetadas geralmente se queixam de sangramentos nasais frequentes e de pequenos sangramentos manchados na face.
Se houver suspeita de doença de Osler, diagnósticos adequados devem ser realizados, uma vez que a vasodilatação também pode ocorrer em órgãos vitais ou com bom suprimento sanguíneo, como pulmões, cérebro ou fígado, nos quais o sangramento de um vaso rompido é perigoso.
Você pode encontrar mais sobre este tópico em nosso site Doença de Osler
Doença de Recklinghausen
A neurofibromatose tipo 1 - ou doença de Recklinghausen - é uma doença genética em que os afetados freqüentemente desenvolvem tumores nas células do revestimento do nervo. Os tumores que se desenvolvem podem ser benignos e malignos e aparecem em uma idade jovem.
Os tumores típicos, entretanto, são neurofibromas benignos: consistem em células que revestem e isolam o nervo como um cabo elétrico, assim como o tecido conjuntivo circundante. Eles são benignos, isto é, tumores que não se propagam e crescem lentamente.
No entanto, a cirurgia para remover os neurofibromas pode ser difícil, pois geralmente eles estão firmemente fixados ao nervo e o nervo correspondente deve ser removido. No entanto, essa é a única opção de tratamento para o neurofibroma sintomático, pois a terapia causal para essa doença hereditária não é possível.
Você pode encontrar mais sobre este tópico em nosso site Neurofibromatose tipo 1
Distrofia muscular
O termo distrofia muscular descreve um grupo de doenças hereditárias nas quais certos componentes musculares não podem ou não podem ser montados corretamente pelas células do corpo. Como resultado, as pessoas afetadas geralmente desenvolvem fraqueza muscular já na infância e adolescência, o que pode resultar em perda de massa muscular, restrições de movimento e até mesmo incapacidades físicas.
Se houver suspeita de distrofia muscular, os valores sanguíneos devem ser determinados primeiro. Se os valores corresponderem ao diagnóstico suspeito, uma biópsia muscular ainda pode ser realizada: Uma pequena amostra de tecido é retirada do músculo, que é então examinada microscopicamente em busca de defeitos celulares. Um exame genético também é possível para estabelecer o diagnóstico, uma vez que as localizações genéticas correspondentes costumam ser conhecidas para as várias formas de distrofia muscular e teriam que ser alteradas. Uma terapia causal para distrofias musculares não é conhecida.
Você pode encontrar mais sobre este tópico em nosso site Distrofia muscular
Xeroderma pigmentoso
O xeroderma pigmentoso é uma doença hereditária rara na qual certas enzimas da pele da pessoa afetada não atuam. Essas enzimas normalmente cuidam do reparo no DNA, que pode ser danificado pela luz solar ou pela luz UVB contida. O dano UVB pode causar câncer de pele nas pessoas afetadas, bem como em todas as outras pessoas, mas com o Xeroderma Pigmentosum o processo é acelerado pela falta de mecanismos de reparo. Como resultado, as pessoas afetadas desenvolvem formas graves de câncer de pele na infância e na adolescência e após uma curta exposição ao sol.
Uma terapia causal ainda não é possível. As pessoas afetadas devem evitar a luz solar por toda a vida, razão pela qual o apelido de "crianças ao luar" se estabeleceu para as pessoas afetadas (às vezes muito jovens). Além disso, essas pessoas devem ser supervisionadas por um dermatologista para exames regulares de câncer de pele, a fim de remover imediatamente o câncer de pele recém-desenvolvido. Se essas medidas forem seguidas estritamente, a expectativa de vida de uma pessoa com xeroderma pigmentoso é aproximadamente a mesma de uma pessoa não afetada.
Você pode encontrar mais sobre esta doença em nosso site Xeroderma pigmentoso
Síndrome de Lynch
A síndrome de Lynch é uma alteração no DNA que causa uma enzima defeituosa nas células do corpo.Nas pessoas afetadas, um certo mecanismo é defeituoso, que de outra forma deveria proteger as células da degeneração, ou seja, crescimento descontrolado - pessoas com síndrome de Lynch, portanto, têm um risco muito maior de desenvolver câncer.
O câncer de cólon freqüentemente ocorre porque as células se dividem naturalmente aqui de qualquer maneira e os erros na programação do crescimento e da morte de uma célula tornam-se aparentes mais rapidamente. As pessoas afetadas frequentemente desenvolvem um tumor no intestino grosso em uma idade incomumente jovem, ou seja, antes dos 50 anos, que é então chamado de HNPCC (câncer de cólon não-pólipo hereditário). No entanto, nem todo mundo que tem a composição genética da síndrome de Lynch desenvolverá câncer de cólon. Por outro lado, outros órgãos também podem desenvolver tumor, uma vez que as predisposições genéticas que favorecem o desenvolvimento de um tumor estão presentes em todas as células do corpo. Verificações regulares e exames preventivos são, portanto, necessários para as pessoas afetadas pela síndrome de Lynch, a fim de tratar adequadamente os tumores que se desenvolvem em um estágio inicial.
Você pode encontrar mais sobre este tópico em nosso site Síndrome de Lynch